Trajectory differentially expression
library(dyno)
library(tidyverse)Compared to differential expression between clusters of cells, defining differential expression on trajectories is not so straightforward. What constitutes a trajectory differentially expressed gene?
- A gene that is uniquely expressed in a particular branch?
- A gene that changes at a branching point?
- A gene that changes along pseudotime?
- …?
dynfeature is a package that allows you to find these different kinds of differential expression in a trajectory. It first defines a particular variable that needs to be predicted (for example, whether a cell is present in a branch or not), and tries to predict this variable based on the expression in different cells. It then ranks each feature based on their predictive capability, and based on this ranking you can select differentially expressed genes.
Depending on what variable is predicted, you get a different ranking. This simply depends on what kind of features you are interested in:
A global overview of the most predictive genes
If you just want to see features that change anywhere in the trajectory, you can use `dynfeature::
model <- dyntoy::generate_dataset(model = dyntoy::model_bifurcating(), num_features = 500)overall_feature_importances <- dynfeature::calculate_overall_feature_importance(model)
features <- overall_feature_importances %>%
top_n(40, importance) %>%
pull(feature_id)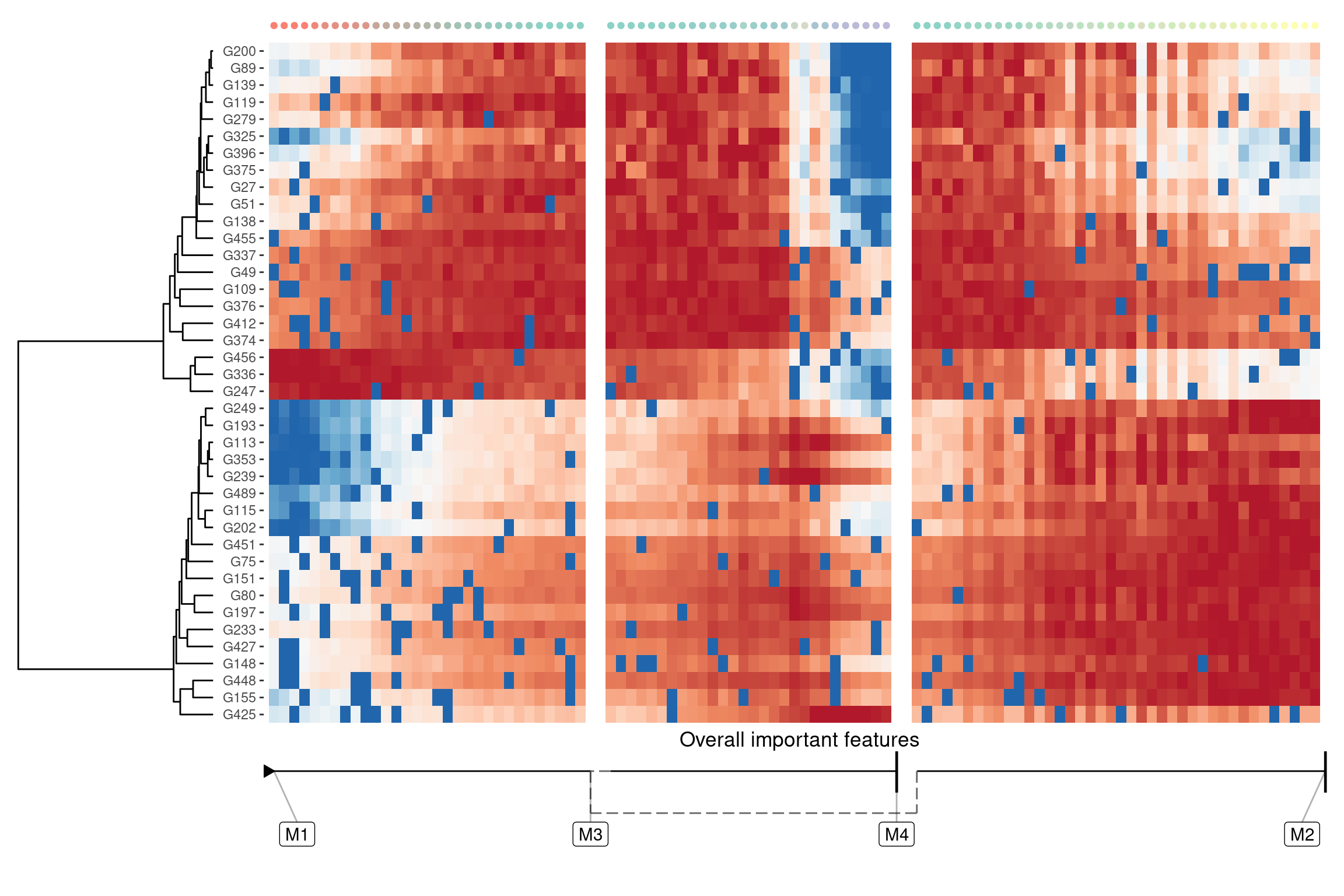
Lineage/branch markers
We can also extract features that are specifically upregulated or downregulated in a specific branch:
branch_feature_importance <- calculate_branch_feature_importance(model)
features <- branch_feature_importance %>%
filter(to == "M4") %>%
top_n(20, importance) %>%
pull(feature_id)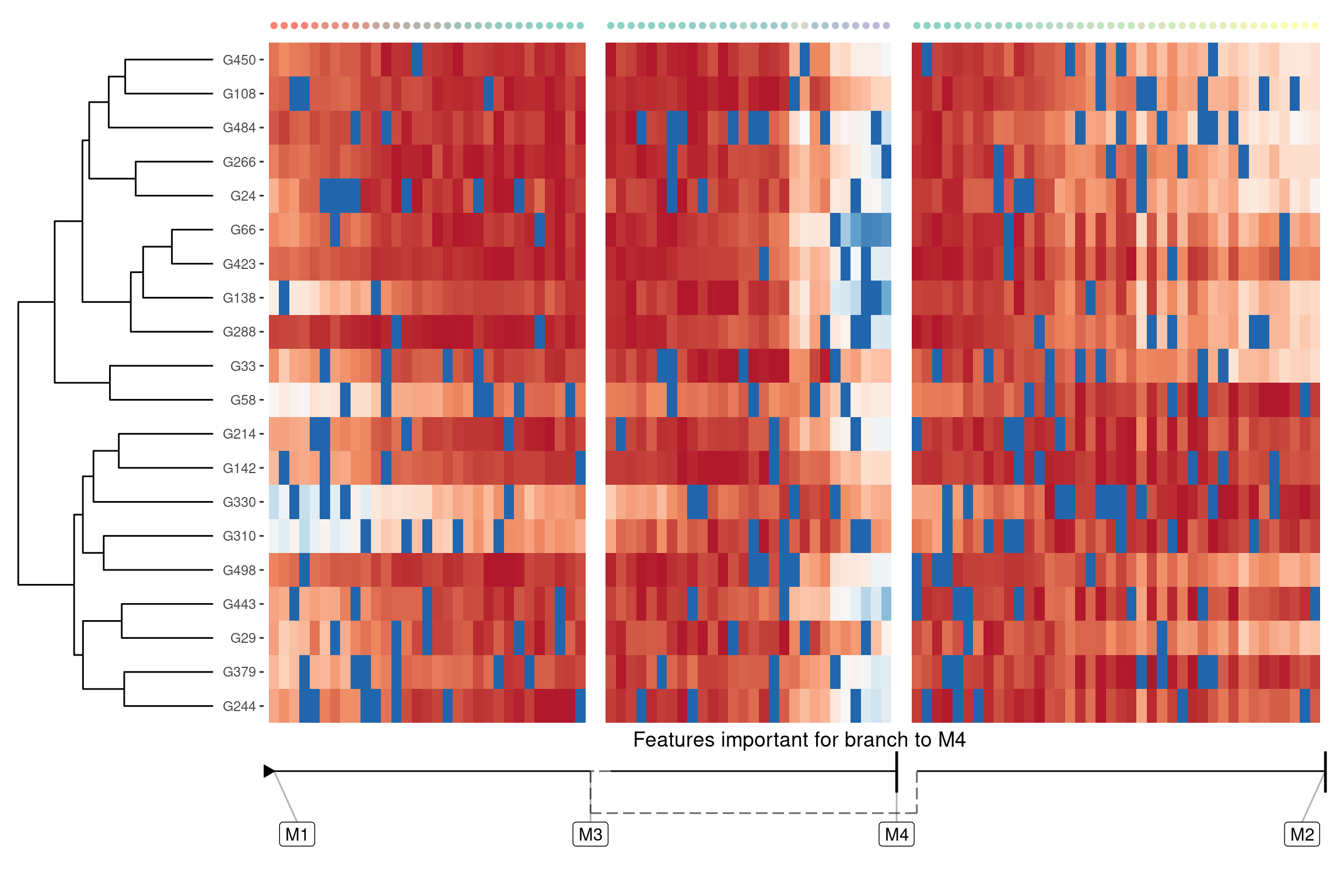
Genes important at bifurcation points
We can also extract features which change at the branching point
branching_milestone <- "M3"
branch_feature_importance <- calculate_branching_point_feature_importance(model, milestones_oi = branching_milestone)
features <- branch_feature_importance %>% top_n(20, importance) %>% pull(feature_id)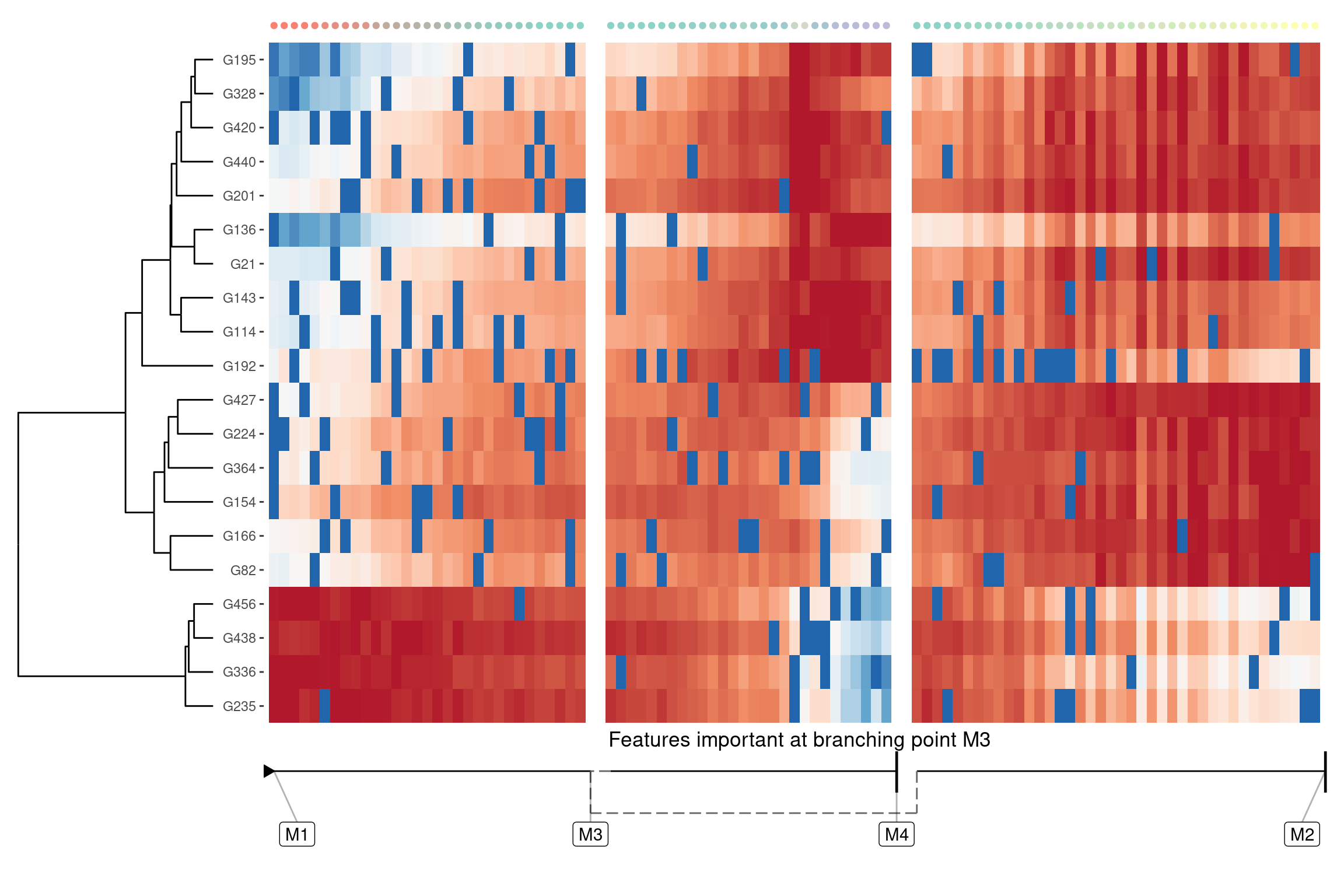
Current limitations
While dynfeature is useful to rank the features according to the strength of trajectory differential expression, they do not provide a statistical ground to find features which are significantly differentially expressed.